
This was my last week doing my internship at ASU with assistant professor Matthias Heyden and his team. This week I was able to work a lot on the super computer and continued to learn the bash scripting language through the terminal.
On Monday I was able to set up my account on the super computer. Every account has their own partitioned storage directory called scratch which is where I held all the files and scripts for the simulations that I was going to run. My main goal by the end of the week was to run a 100 nanosecond simulation of a lysozyme using GROMACS, an open source process that allows for easy compilations. Once I got into my scratch directory, one of the people on Matthias’ team sent me the appropriate files to run the simulation. There were many bugs that weren’t allowing the simulation to run on my account and so for much of Monday I was working on resolving those bugs.
During Tuesday, most of the bugs were resolved. This allowed me to get a partial way into the simulation process until another bug occurred. The same person who helped me transfer the simulation files onto my account worked on figuring out why some files weren’t being created. While he did that, I was able to learn more about different force fields, which are calculation algorithms for the physics and interactions of atoms. I also spent time researching the modes of molecules so I would better understand what I was really simulating. Modes in a molecule are considered to be the variety of ways that it can bend and stretch (vibrate). For example, if you have a carbon dioxide molecule that looks like this -> O=C=O, it can stretch asymmetrically like this, -> OC==O or O==CO. These modes affect the interactions that it has with other molecules and atoms because of the change in position that the vibrations cause. By the end of the day, all the bugs were resolved and so I could continue with running the simulation on Wednesday.
Wednesday I began the day off with a meeting that involved the different people in Matthias’ group. They each showed what they have been working on which included a lot of interesting stuff. For example, one of the members on the team used a monte carlo simulation to accurately determine the positioning of proteins. Once the meeting was done, I was told to look over all the files and bash code so I could better understand what was happening under the hood. I wasn’t to familiar with bash scripting, so I quickly researched how the language functioned which didn’t take to long. There were about 8 bash files and the rest were either .mdp or random miscellaneous files. A .mdp file is a file that specifies meta dynamics parameters on a system which means the file contains the time for a simualtion, the modes, or any normalizations. After looking at the .mdp files, I went into the bash scripts which weren’t too hard to understand. The only lines of the scripts I didn’t fully understand were the GROMACS lines because of the size of the library.
Thursday I was able to start running the simulation on my scratch directory. Luckily, there were no errors that occurred so all I had to do was wait for the simulation to finish. During the time that I was waiting, I tried building my own physics simulation in python using an Open GL library. I applied gravity to any new objects, energy loss, and the hardest part were the collisions. After a while of tinkering and looking at different code implementations that I could use to most effectively calculate efficient collisions, I finally got the different objects in the system to collide with a good time complexity.
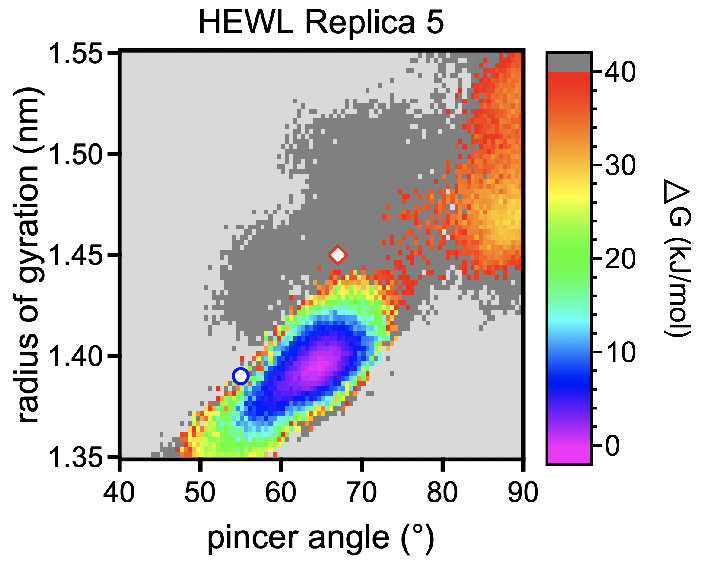
Finally on Friday, I was able to look at my simulation and analyze the results. I moved the simulation file into my own local home directory and then booted up VMD (VMD is just a 3d rendering application). I was able to visualize the different domains of the lysozyme and test out how the protein position calculations changed based on the mode that was applied to the system. I also saw a plot that showed the different amounts of free energy in the protein at different vibrational tendencies and wider angles. The simulation produced 1000 frames which gave me a good understanding of how the protein changes its shape based on free energy within the system.
When I was not on campus, I connected with my roommate with lots of video games and tv shows.
I am so happy that I had the opportunity to learn about computational chemistry at the tail end of my summer, and I thank Matthias Heyden and his team so much for giving me helpful insight into their field!
There are no comments published yet.